
Izmailov, S. A.; Podkorytov, I. S.; Skrynnikov, N. R. Simple MD-Based Model for Oxidative Folding of Peptides and Proteins. Scientific Reports 2017, 7 (1).
DOI: 10.1038/s41598-017-09229-7.
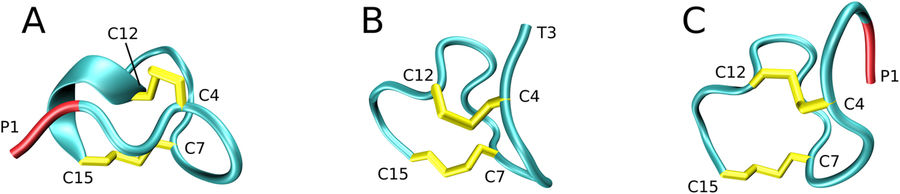
Significant strides have been recently made to fold peptides and small proteins in silico using MD simulations. However, facilities are currently lacking to include disulfide bonding in the MD models of protein folding. To address this problem, we have developed a simple empirical protocol to model formation of disulfides, which is perturbation-free, retains the same speed as conventional MD simulations and allows one to control the reaction rate. The new protocol has been tested on 15-aminoacid peptide guanylin containing four cysteine residues; the net simulation time using Amber ff14SB force field was 61 μs. The resulting isomer distribution is in qualitative agreement with experiment, suggesting that oxidative folding of guanylin in vitro occurs under kinetic control. The highly stable conformation of the so-called isomer 2(B) has been obtained for full-length guanylin, which is significantly different from the poorly ordered structure of the truncated peptide PDB ID 1GNB. In addition, we have simulated oxidative folding of guanylin within the 94-aminoacid prohormone proguanylin. The obtained structure is in good agreement with the NMR coordinates 1O8R. The proposed modeling strategy can help to explore certain fundamental aspects of protein folding and is potentially relevant for manufacturing of synthetic peptides and recombinant proteins.
← PreviousТэги: Измайлов, Подкорытов, Скрынников