
Ning Liu, Oleg Mikhailovskii, Nikolai R. Skrynnikovb, and Yi Xue
https://journals.iucr.org/m/issues/2023/01/00/lz5062/index.html
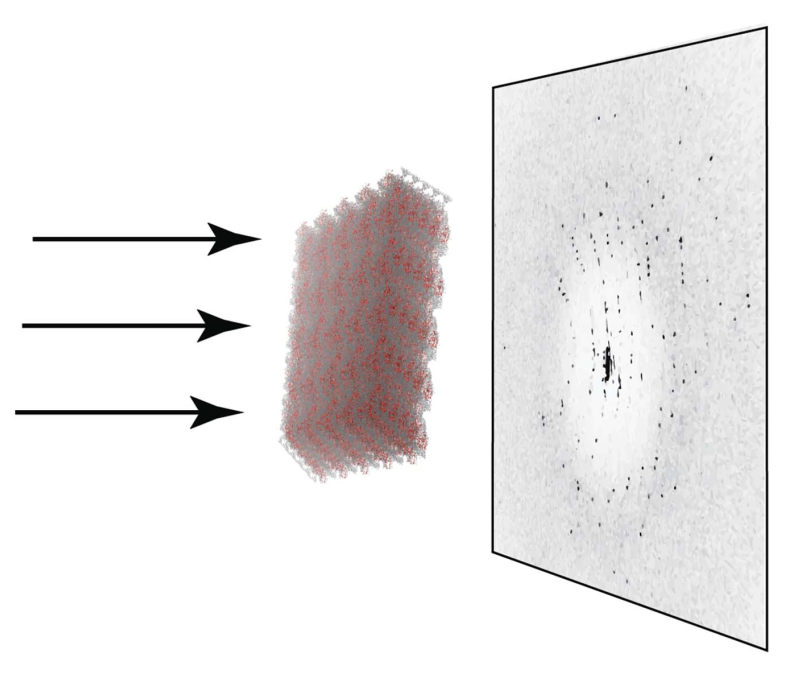
A molecular dynamics (MD)-based pipeline has been designed and implemented to emulate the entire process of collecting diffraction photographs and calculating crystallographic structures of proteins from them. Using a structure of lysozyme solved in-house, a supercell comprising 125 (5 × 5 × 5) crystal unit cells containing a total of 1000 protein molecules and explicit interstitial solvent was constructed. For this system, two 300 ns MD trajectories at 298 and 250 K were recorded. A series of snapshots from these trajectories were then used to simulate a fully realistic set of diffraction photographs, which were further fed into the standard pipeline for structure determination. The resulting structures show very good agreement with the underlying MD model not only in terms of coordinates but also in terms of B factors; they are also consistent with the original experimental structure. The developed methodology should find a range of applications, such as optimizing refinement protocols to solve crystal structures and extracting dynamics information from diffraction data or diffuse scattering.
← PreviousТэги: Mikhailovskii, Ning Liu, Skrynnikov, Yi Xue